
Autosomno dominantna bolest policistnih bubrega
| Autosomna bolest policistnih bubrega (Autosomna dominantna PKD) (Adultno pojavna PKD) (Mnogocistasta autosomna bolest bubrega)  | |
|---|---|
| Klasifikacija i vanjski resursi | |
| ICD-10 | Q61 |
| ICD-9 | 753.1 |
| OMIM | 601313 OMIM: 173910 |
| DiseasesDB | 10262 |
| MedlinePlus | 000502 |
| eMedicine | radio/68 |
| MeSH | D016891 |
Autosomna dominantna policistna bubrežna bolest (ADPKD) je najrasprostranjenija, potencijalno smrtonosna, monogenski ljudski poremećaj.[1] Povezana je s velikom međuporodičnom i unutarporodičnom varijabilnošću, što se u velikoj mjeri može objasniti genetičkom heterogenošću i modifikacijom djelovanja gena. Također je najčešća od nasljednih cistastih bolesti bubrega – grupe poremećaja sa povezanom, ali različitom patogenezom, karakteriziranom razvojem bubrežnim cistama i različitim vanbubrežnim manifestacijama, koje u slučaju ADPKD uključuju ciste u drugim organima, kao što su jetra, sjemeni mjehurić, gušterača i arahnoidna membrana, kao i druge abnormalnosti, poput intrakranijalnih aneurizmi i dolihoektazijaza, korijen aorte dilatacija i aneurizme, prolaps mitralnog zaliska i hernije trbušnog zida.[1][2][3] Preko 50% pacijenata sa ADPKD-om na kraju razvije završni stadij bubrežne bolesti i treba dijalisu ili transblantaciju bubrega.[1][4] Procjenjuje se da ADPKD utiče na najmanje jednu na svakih 1.000 osoba širom svijeta, čineći ovu bolest najčešćim nasljednim poremećajem bubrega s dijagnosticiranom prevalencijom 1:2000 i incidencijom 1:3000–1: 8000 u globalnim razmjerima.[5][6][7][8][9]
Znakovi i simptomi
[uredi | uredi izvor]- Akutni bol u krstima
- Krv u mokraći
- Loptasti bubrezi
- Subarahnoidno krvarenje (bobičasta aneurizma)
- Hipertenzija
- Povezane ciste jetre
- Uremija zbog otkazivanja bubrega
- Anemija zbog hronične bubrežne bolesti
- Povećana sekrecija eritrocita ili eritropoetina
Genetika
[uredi | uredi izvor]ADPKD je genetički heterogena bolest pod kontrolom sa dva identificirana gena: PKD1 (regija hromozoma 16p13.3; oko 85% slučajeva) i PKD2 (4q21; oko 15% slučajeva).[1] Nekoliko genetičkih mehanizama vjerovatno doprinosi fenotipskom iispoljavanju bolesti.[1] Iako postoje dokazi o mehanizmu dva pogotka (zametna linija i somatska inaktivacija dva alela PKD) koji objašnjavaju žarišni razvoj bubrežnih i jetrenih cista,[10][11] a haploinsuficijencija će vjerovatnije objasniti vaskularne manifestacije bolesti.[12][13] Pored toga, novi modeli miša, homozigotni za PKD1 hipomorfne alele 22 i 23 i demonstiraju povećane proliferacije bubrežnih epitelnih ćelija kod PKD2 +/– miševa sugeriraju da mehanizmi koji nisu hipoteza o dva pogotka također doprinose fenotipu sa cistama.[1]
U ADPKD-u se javlja velika međuporodična i unutarporodična varijabilnost.[1] Većina osoba s mutacijom PKD1 ima zatajenje bubrega u dobi od 70 godina, dok više od 50% osoba sa mutacijom PKD2 imaju odgovarajuću bubrežnu funkciju u toj dobi (srednja dob početka završnog stadija bubrega: 54,3 godine sa PKD1 ; 74,0 godina sa PKD2).[14]
Značajna unutarporodična varijabilnost uočena u ozbiljnim bubrežnim i vanbubrežnim manifestacijama ukazuje na genetičke i ekološke faktore koji mogu uticati na ishod ADPKD, a rezultati analize varijabilnosti bubrežne funkcije između monozigotnih blizanaca i braće i sestara podržavaju ulogu genetičkih modifikatora kod ove bolesti.[1][15] Procjenjuje se da bi 43–78% varijanse u dobi prema ESRD moglo biti posljedica nasljednih modificirajućih faktora,[16][17] sa roditeljima koji imaju vjerovatnije kao djeca da pokažu ozbiljniju bolest u istraživanjima parova roditelj-dijete.[1][18]
Patofiziologija
[uredi | uredi izvor]U mnogih pacijenata sa ADPKD, bubrežna disfunkcija nije klinički očigledna do 40. ili 50. godine života.[4] Međutim, sve veći broj dokaza sugerira da stvaranje bubrežnih cista započinje in utero.[19] Ciste se u početku formiraju kao male dilatacije u bubrežnim tubulima, koje se potom šire i formiraju šupljine ispunjene tečnošću različitih veličina.[19] Faktori za koje se sugerira da vode do cistogeneze uključuju mutaciju zametne linije u jednom od alelnih gena policistina, somatski drugi pogodak koji dovodi do gubitka normalnog alela i treći pogodak, koji može biti sve što pokreće proliferaciju ćelija, dovodeći do širenja tubula.[19] U progresiji bolesti, kontinuirano širenje tubula putem povećanja proliferaciju ćelija, lučenje tečnosti i odvajanje od roditeljskih tubula dovodi do stvaranja cista.[20][21]
ADPKD, zajedno sa mnogim drugim bolestima koje se javljaju s bubrežnim cistama, mogu se klasificirati u porodicu bolesti poznatih kao ciliopatije.[22] Epitelne ćelije bubrežnih tubula, uključujući sve segmente nefrona i sabirne kanale (s izuzetkom interkaliranih ćelija) pokazuju prisustvo jedne primarne apikalne treplje.[23] Policistin-1, protein kodiran genom PKD1, prisutan je na tim trepljama i smatra se da osjeća protok, svojim velikim vanćelijskim domenima, aktivirajući kalcijeve kanale povezane sa policistinom-2, proizvode gena PKD2,[24] kao rezultat genetičke postavke ADPKD, kao što je objašnjeno u pododjeljku genetika.
Proliferacija epitelnih ćelija i lučenje tečnosti koje dovode do cistogeneze dvije su karakteristike ADPKD.[25] Tokom ranih faza cistogeneze, ciste su vezane za roditeljske bubrežne tubule i derivat glomerulskog filtrata ulazi u ciste.[19] Kada se te ciste prošire na otprilike 2 mm u promjeru, cista se zatvori iz roditeljskog tubula i nakon toga tečnost može ući u ciste samo putem transepitelne sekrecije, za koju se pak predlaže povećanje zbog sekundarnih efekata povećane unutarćelijske koncentracije cikličnog AMP-a (cAMP).[19]
Klinički, podmuklo povećanje broja i veličine bubrežnih cista prevodi se kao progresivno povećanje volumena bubrega.[1][19] Studije koje su vodili stručnjaci Mayo Clinic utvrdile su da je ukupni volumen bubrega (TKV) u velikoj kohorti pacijenata sa ADPKD bio 1060 ± 642 ml, sa srednjim porastom od 204 ml tokom tri godine ili 5,27% godišnje u prirodnom toku bolesti, između ostalih važnih, novih otkrića koja su po prvi put opsežno proučavana.[26]

Dijagnoza
[uredi | uredi izvor]Obično se dijagnoza ADPKD u početku provodi snimanjem bubrega pomoću ultrazvuka, CT-skeniranjem ili putem MRI.[27] Međutim, molekulska dijagnostika može biti potrebna u sljedećim situacijama:
- 1: kada je kod mladih osoba potrebna definitivna dijagnoza, kao što je potencijalni donor u živoj porodici u pogođenoj porodici s dvosmislenim slikovnim podacima;[27]
- 2: kod pacijenata sa negativnom porodičnom anamnezom ADPKD, zbog potencijalnog fenotipskog preklapanja sa nekoliko drugih cistnih bolesti bubrega;[27]
- 3: u porodicama pogođenim ranom pojavom policistne bolesti bubrega, jer u ovom slučaju mogu biti uključeni hipomorfni aleli i/ili oligogensko nasljeđivanje;[27][28] i
- 4: kod pacijenata koji traže genetičko savjetovanje, posebno kod parova koji žele predimplantaciju genetičku dijagnozu.[27][29]
Dijagnostički nalazi su sa velikim ehogenim bubrezima, bez izraženih makroskopskih cista kod novorođenčeta/djeteta s 50% rizika za ADPKD. U nedostatku porodične anamneze ADPKD, prisustvo obostranog proširenja bubrega i cista, sa ili bez prisustva jetrenih cista, i odsustvo drugih manifestacija koje ukazuju na različitu bubrežnu cistnu bolest daju pretpostavku, ali ne i definitivan dokaz za dijagnozu. U nekim slučajevima, intrakranijske aneurizme mogu biti pridruženi znak ADPKD-a, a skrining se može preporučiti pacijentima sa porodičnom anamnezom intrakranijske aneurizme.[30]
Klinički je dostupno molekulsko genetičko testiranje pomoću analiza povezanosti ili izravni pregled mutacija; međutim, genetička heterogenost je značajna komplikacija za molekulsko genetičko testiranje. Ponekad treba testirati relativno veliki broj pogođenih članova porodice, kako bi se utvrdilo koji je od dva moguća gena odgovoran u svakoj porodici. Velika veličina i složenost gena PKD1 i PKD2, kao i označena alelna heterogenost, predstavljaju prepreke za molekulsko ispitivanje direktnim DNK analizama. Osetljivost testiranja je gotovo 100% za sve pacijente sa ADPKD koji su stariji od 30 godina i za mlađe pacijente sa mutacijama PKD1; ovi su kriteriji osjetljivi samo na 67% za pacijente sa mutacijama PKD2, koji su mlađi od 30 godina.
-
 Policistni bubreg odrasle osobe
Policistni bubreg odrasle osobe -
 Dijagram autosomno dominantne policistne bolesti sa uloškom normalnog bubrega za usporedbu
Dijagram autosomno dominantne policistne bolesti sa uloškom normalnog bubrega za usporedbu -
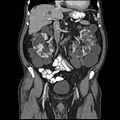 CT abdomena odrasle osobe s autosomno dominantnom policistnom bolešću bubrega: Opsežno stvaranje cista je na oba bubrega, s nekoliko cista u jetri, također. (koronaska ravan)
CT abdomena odrasle osobe s autosomno dominantnom policistnom bolešću bubrega: Opsežno stvaranje cista je na oba bubrega, s nekoliko cista u jetri, također. (koronaska ravan)



Liječenje
[uredi | uredi izvor]Danas se jedini dostupni klinički/farmakološki tretman za ADPKD sastoji u smanjenju brzine dobijanja ukupne zapremine bubrega (TKV) akvareticima (tj. Tolvaptanom), što može ublažiti bol, dok pacijentima pruža bolji kvalitet života tokom više od tri godine. Nakon ovog perioda, pacijenti mogu ponovno započeti dobivanje TKV-a brzinom predtretmana i na kraju će možda morati proći dijalizu i transplantaciju bubrega. Načini palijativnog liječenja uključuju simptomatske lijekove (nonopioidne i opioidne analgetike) za trbušne/retroperitoneumske bolove. Prije pojave akvauretskih lijekova, jedina opcija za bol koji je otporan na analgetike, bili su jednostavni ili složeni hirurški zahvati (tj. aspiracija bubrežne ciste, dekortizacija ciste, bubrežna denervacija i nefrektomija), što može rezultirati komplikacijama svojstvenim operaciji.
U liječnju mogu se primijeniti:
- Akvauretski lijekovi
- Analgetički lijekovi
- Aspiracija bubrežne ciste
- Laparoskopska dekortikacija ciste
- Nefrektomija (uklanjanje bubrega)
- Dijaliza
- Transplantacija bubrega
Prognoza
[uredi | uredi izvor]Kod pacijenata sa ADPKD, postepeni razvoj i širenje cista rezultira povećanjem bubrega, a tokom bolesti stopa glomerulske filtracije ostaje normalna desetljećima prije nego što funkcija bubrega počne postupno pogoršavati, što predviđa rani težak ishod.[31] Studije CRISP, spomenute u gornjem odjeljku liječenje, doprinijele je stvaranju snažne argumentacije koja podržava prognostičku vrijednost ukupne zapremine bubrega (TKV) u ADPKD; TKV (procjenjuje se pomoću MRI) kontinuirano raste i veća stopa proširenja bubrega korelira s ubrzanim padom GFR-a, dok TKV (HtTKV) prilagođen visini pacijenta ≥600 ml / m predviđa razvoj hronične bolesti bubrega u fazi 3 unutar 8 godina.
Pored TKV i HtTKV, procijenjena brzine glomerulske filtracije (eGFR) također je privremeno korištena za predviđanje napredovanja ADPKD. Nakon analize CT ili MRI snimaka 590 pacijenata sa ADPKD-om koji su liječeni u Mayo Clinic, Irazabal i njegovi kolege razvili su sistem klasifikacije zasnovan na slikama kako bi predvidjeli stopu pada eGFR-a kod pacijenata sa ADPKD-om.[32] Ovim prognostičkom metodom pacijenti su podijeljeni u pet potklasa procijenjenih stopa rasta bubrega prema rasponima HtTKV specifičnih za dob (1A, <1,5%; 1B, 1,5-3,0%; 1C, 3,0-4,5%; 1D, 4,5-6,0% i 1E,> 6,0%) kako je navedeno u studiji CRISP. Pad eGFR-a tokom godina nakon početnog mjerenja TKV značajno se razlikuje između svih pet podtlasa pacijenata, a oni iz potklase 1E imaju najbrži pad.[31] Neki od najčešćih uzroka smrti kod pacijenata sa ADPKD su razne infekcije (25%), puknuće bobičastih aneurizmi (15%) ili koronska/hipertenzivna bolest srca (40%).[33]
Reference
[uredi | uredi izvor]- ^ a b c d e f g h i j Torres VE, Harris PC, Pirson Y (2007). "Autosomal dominant polycystic kidney disease". Lancet. 369 (9569): 1287–1301. doi:10.1016/S0140-6736(07)60601-1. PMID 17434405. S2CID 1700992.
- ^ Dalgaard OZ (1957). "Bilateral polycystic disease of the kidneys; a follow-up of two hundred and eighty-four patients and their families". Acta Med. Scand. Suppl. 328: 1–255. PMID 13469269.
- ^ Torres, Vicente; Harris, Peter C (20. 5. 2009). "Autosomal dominant polycystic kidney disease: the last 3 years". Kidney International. 76 (2): 149–168. doi:10.1038/ki.2009.128. PMC 2812475. PMID 19455193.
- ^ a b Grantham JJ (2008). "Clinical practice. Autosomal dominant polycystic kidney disease". N. Engl. J. Med. 359 (14): 1477–1485. doi:10.1056/NEJMcp0804458. PMID 18832246.; Reprinted in Niemczyk M, Niemczyk S, Paczek L (2009). "Autosomal dominant polycystic kidney disease and transplantation". Ann Transplant. 14 (4): 86–90. PMC 2843931. PMID 20009161.
- ^ Muto S, Kawano H, Higashihara E, Narita I, Ubara Y, Matsuzaki T, Ouyang J, Torres VE, Horie S (2015). "The effect of tolvaptan on autosomal dominant polycystic kidney disease patients: a subgroup analysis of the Japanese patient subset from TEMPO 3:4 trial". Clin Exp Nephrol. 19 (5): 867–877. doi:10.1007/s10157-015-1086-2. PMID 25663351. S2CID 12124902.
- ^ Higashihara E, Nutahara K, Kojima M, Tamakoshi A, Yoshiyuki O, Sakai H, Kurokawa K (1998). "Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan". Nephron. 80 (4): 421–427. doi:10.1159/000045214. PMID 9832641. S2CID 22124996.
- ^ Levy M, Feingold J (2000). "Estimating prevalence in single-gene kidney diseases progressing to renal failure". Kidney Int. 58 (3): 925–943. doi:10.1046/j.1523-1755.2000.00250.x. PMID 10972657.
- ^ Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS (2012). "Tolvaptan in patients with autosomal dominant polycystic kidney disease". N. Engl. J. Med. 367 (25): 2407–2418. doi:10.1056/NEJMoa1205511. PMC 3760207. PMID 23121377.
- ^ Cornec-Le Gall E, Le Meur Y (2014). "Autosomal dominant polycystic kidney disease: is the treatment for tomorrow?". Nephrol. Ther. 10 (6): 433–440. doi:10.1016/j.nephro.2014.03.003. PMID 25086476.
- ^ Torra R, Badenas C, San Millán JL, Pérez-Oller L, Estivill X, Darnell A (1999). "A loss-of-function model for cystogenesis in human autosomal dominant polycystic kidney disease type 2". Am. J. Hum. Genet. 65 (2): 345–352. doi:10.1086/302501. PMC 1377933. PMID 10417277.
- ^ Watnick TJ, Torres VE, Gandolph MA, Qian F, Onuchic LF, Klinger KW, Landes G, Germino GG (1998). "Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease". Mol. Cell. 2 (2): 247–251. doi:10.1016/s1097-2765(00)80135-5. PMID 9734362.
- ^ Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, Harris PC, Torres VE, Sieck GC (2003). "PKD2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells". Mol. Cell. 12 (15): 1875–1880. doi:10.1093/hmg/ddg190. PMID 12874107.
- ^ Gao Z, Joseph E, Ruden DM, Lu X (2004). "Drosophila Pkd2 is haploid-insufficient for mediating optimal smooth muscle contractility". J. Biol. Chem. 279 (14): 14225–14231. doi:10.1074/jbc.M312223200. PMID 14732716.
- ^ Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D (1999). "Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group". Lancet. 353 (9147): 103–107. doi:10.1016/s0140-6736(98)03495-3. PMID 10023895. S2CID 30757096.
- ^ Persu A, Duyme M, Pirson Y, Lens XM, Messiaen T, Breuning MH, Chauveau D, Levy M, Grünfeld JP, Devuyst O (2004). "Comparison between siblings and twins supports a role for modifier genes in ADPKD". Kidney Int. 66 (6): 2132–2136. doi:10.1111/j.1523-1755.2004.66003.x. PMID 15569302.
- ^ Fain PR, McFann KK, Taylor MR, Tison M, Johnson AM, Reed B, Schrier RW (2005). "Modifier genes play a significant role in the phenotypic expression of PKD1". Kidney Int. 67 (4): 1256–1267. doi:10.1111/j.1523-1755.2005.00203.x. PMID 15780078.
- ^ Paterson AD, Magistroni R, He N, Wang K, Johnson A, Fain PR, Dicks E, Parfrey P, St George-Hyslop P, Pei Y (2005). "Progressive loss of renal function is an age-dependent heritable trait in type 1 autosomal dominant polycystic kidney disease". J. Am. Soc. Nephrol. 16 (3): 755–762. doi:10.1681/ASN.2004090758. PMID 15677307.
- ^ Geberth S, Ritz E, Zeier M, Stier E (1995). "Anticipation of age at renal death in autosomal dominant polycystic kidney disease (ADPKD)?". Nephrol. Dial. Transplant. 10 (9): 1603–1606. PMID 8559477.
- ^ a b c d e f Paul BM, Vanden Heuvel GB (2014). "Kidney: polycystic kidney disease". Wiley Interdiscip. Rev. Dev. Biol. 3 (6): 465–487. doi:10.1002/wdev.152. PMC 4423807. PMID 25186187.
- ^ Igarashi P, Somlo S (2002). "Genetics and pathogenesis of polycystic kidney disease". J. Am. Soc. Nephrol. 13 (9): 2384–2398. doi:10.1097/01.asn.0000028643.17901.42. PMID 12191984.
- ^ Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP (2002). "Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins". J. Biol. Chem. 277 (22): 19566–19572. doi:10.1074/jbc.M201875200. PMID 11912216.
- ^ Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (2009). "The primary cilium as a complex signaling center". Curr. Biol. 19 (13): R526–R535. doi:10.1016/j.cub.2009.05.025. PMC 2814769. PMID 19602418.
- ^ Reed BY, McFann K, Bekheirnia MR, Nobakhthaghighi N, Masoumi A, Johnson AM, Shamshirsaz AA, Kelleher CL, Schrier RW (2008). "Variation in age at ESRD in autosomal dominant polycystic kidney disease". Am. J. Kidney Dis. 51 (2): 173–183. doi:10.1053/j.ajkd.2007.10.037. PMC 2747334. PMID 18215695.
- ^ Chapin HC, Caplan MJ (2010). "The cell biology of polycystic kidney disease". J. Cell Biol. 191 (4): 701–710. doi:10.1083/jcb.201006173. PMC 2983067. PMID 21079243.
- ^ Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Grantham JJ (2004). "Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells". Kidney Int. 66 (3): 964–973. doi:10.1111/j.1523-1755.2004.00843.x. PMID 15327388.
- ^ Torres VE (2010). "Treatment strategies and clinical trial design in ADPKD". Adv. Chronic Kidney Dis. 17 (2): 190–204. doi:10.1053/j.ackd.2010.01.006. PMC 4127876. PMID 20219622.
- ^ a b c d e Trujillano D, Bullich G, Ossowski S, Ballarín J, Torra R, Estivill X, Ars E (2014). "Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing". Mol. Genet. Genomic Med. 2 (5): 412–421. doi:10.1002/mgg3.82. PMC 4190876. PMID 25333066.
- ^ Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, Hampel T, Pape L, Buske A, Jonsson J, Sarioglu N, Santos A, Ferreira JC, Becker JU, Cremer R, Hoefele J, Benz MR, Weber LT, Buettner R, Zerres K (2011). "Mutations in multiple PKD genes may explain early and severe polycystic kidney disease". J. Am. Soc. Nephrol. 22 (11): 2047–2056. doi:10.1681/ASN.2010101080. PMC 3279997. PMID 22034641.
- ^ Harris PC, Rossetti S (2010). "Molecular diagnostics for autosomal dominant polycystic kidney disease". Nature Reviews Nephrology. 6 (4): 197–206. doi:10.1038/nrneph.2010.18. PMC 4050432. PMID 20177400.
- ^ Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC (2013). "Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms?" (PDF). AJNR Am J Neuroradiol. 35 (1): 3–9. doi:10.3174/ajnr.A3437. PMC 7966475. PMID 23292526. S2CID 5777115.
- ^ a b Cornec-Le Gall E, Le Meur Y (2014). "Polycystic kidney disease: Kidney volume--a crystal ball for ADPKD prognosis?". Nature Reviews Nephrology. 10 (9): 485–486. doi:10.1038/nrneph.2014.132. PMID 25092148. S2CID 22042874.
- ^ Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE (2015). "Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials". J. Am. Soc. Nephrol. 26 (1): 160–172. doi:10.1681/ASN.2013101138. PMC 4279733. PMID 24904092.
- ^ Kumar, Vinay; Abbas, Abul K.; Aster, Jon C. (2014). Robbins and Cotran pathologic basis of disease. Kumar, Vinay, 1944-, Abbas, Abul K.,, Aster, Jon C.,, Perkins, James A. (Ninth izd.). Philadelphia, PA. str. 947. ISBN 9781455726134. OCLC 879416939.
Vanjski linkovi
[uredi | uredi izvor]- https://web.archive.org/web/20110608142128/http://kidney.niddk.nih.gov/kudiseases/pubs/polycystic/index.htm
- https://www.ncbi.nlm.nih.gov/disease/PKD.html
Šablon:Kongenitalne malformacije mokraćnog sistema Šablon:Cistične bolesti